
- Title
-
Zebrafish rbm8a and magoh mutants reveal EJC developmental functions and new 3'UTR intron-containing NMD targets
- Authors
- Gangras, P., Gallagher, T.L., Parthun, M.A., Yi, Z., Patton, R.D., Tietz, K.T., Deans, N.C., Bundschuh, R., Amacher, S.L., Singh, G.
- Source
- Full text @ PLoS Genet.
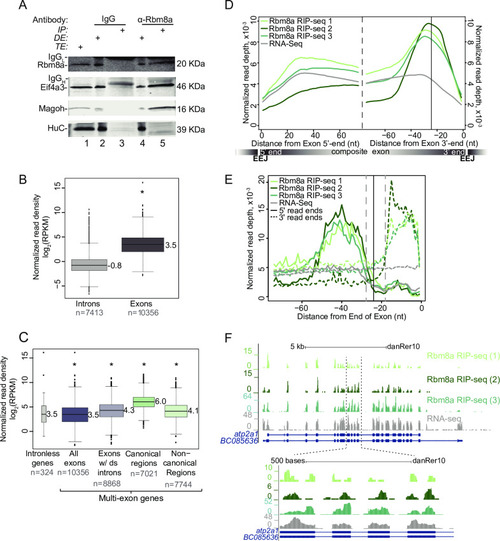
A. Western blot indicating Rbm8a, Eif4a3, Magoh and HuC proteins detected in RNase I-treated zebrafish embryo total extract (TE, lane 1), depleted extract (DE, lanes 2 and 4) immunoprecipitated protein complexes (IP, lanes 3 and 5). Antigens detected in the blot are listed on the left and antibodies used to immunoprecipitate complexes are listed on top. The signal corresponding to the antibody light chain and heavy chain in the IP lanes is indicated by IgGL and IgGH, respectively. B. Boxplots showing the Rbm8a RIP-Seq normalized read densities (reads per kilobase per million, RPKM) in intronic versus exonic genomic regions. Asterisk at the top indicates Wilcoxon test p-value, which is < 10−6. C. Boxplots as in B showing the Rbm8a RIP-Seq normalized read densities (RPKM) in the indicated genomic regions (bottom). Exons with downstream introns include all but last exons. Asterisk at the top indicates Wilcoxon test p-values, which are < 10−6. D. Meta-exon plots showing Rbm8a RIP-Seq and RNA-Seq (indicated on the top left) normalized read depths in a 75 nt region starting from the exon 5′ (left of dashed black line) or 3′ ends (right of dashed black line). Vertical black line: expected canonical EJC binding site (-24 nt) based on human studies. A composite exon with the relative position of exon-exon junctions (EEJ) is diagrammed at the bottom. E. A meta-exon plot of start and end of Rbm8a RIP-Seq or RNA-Seq reads (indicated on the top left; 5′ ends, solid lines; 3′ ends, dotted lines). Vertical black line: canonical EJC site (-24 nt). Gray vertical dashed lines represent boundaries of the minimal EJC occupied site. F. Top: UCSC genome browser screenshots showing read coverage along the |

A. Schematic illustrating the PHENOTYPE:
|
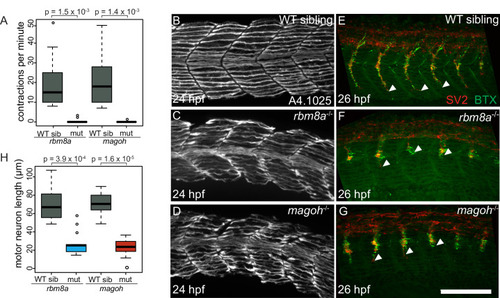
A. Boxplots showing the number of spontaneous contractions per minute measured for the EJC mutant embryos and WT siblings at 24 hpf as indicated on the x-axis. Welch’s t-test p-values are indicated at the top. B-D. Immunofluorescence images showing Myh1 expression in somites 10–14 of WT sibling (B) PHENOTYPE:
|
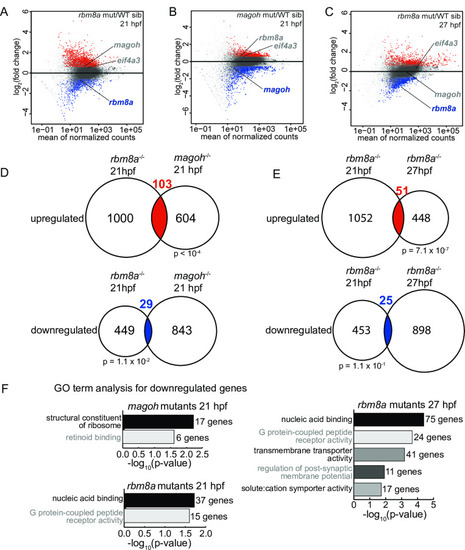
A-C. MA plots (M: log ratio; A: mean average) showing genes that are upregulated (fold change > 1.5 and FDR < 0.05) (red), downregulated (fold change < 1.5 and FDR < 0.05) (blue), or unchanged (gray) in |
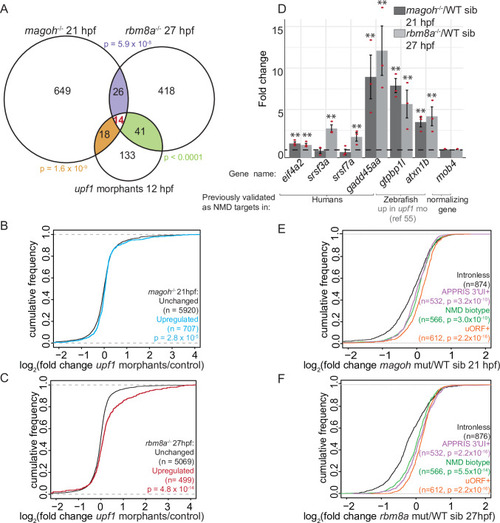
A. Venn diagram showing the overlap of significantly upregulated genes in EJC mutant embryos and EXPRESSION / LABELING:
PHENOTYPE:
|
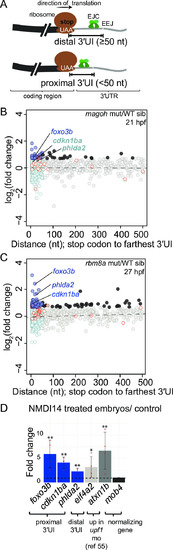
A. Top: Schematic illustrating genes with 3′UTR introns (3′UI) where the distance between the stop codon and the 3′UI is equal to or greater than 50 nts. Such 3′UI are classified as distal. Bottom: Schematic illustrating genes with 3′UI where the distance between the stop codon and 3′UI is less than 50 nts. Such 3′UI are classified as proximal. The ribosome (brown), direction of translation (black arrow), stop codon (‘UAA’ in white), EJC (green), exon-exon junction (EEJ), coding region of mRNA (black) and 3′UI of mRNA (gray) are labeled in the top panel. B. A scatter plot showing gene-level fold change (FC) for transcripts with proximal 3′UI (dark blue: FC > 1.5 and light blue: FC < 1.5) and distal 3′UI (black: FC > 1.5 and gray: FC < 1.5) in |
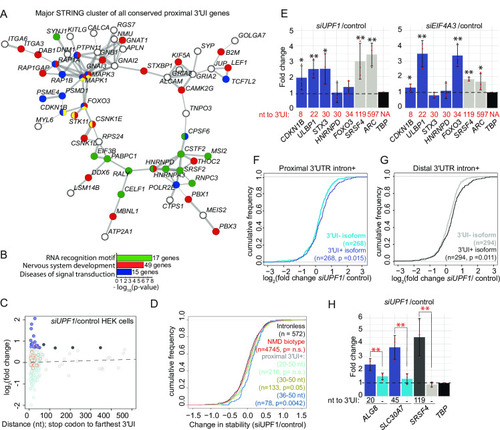
A. A major interaction cluster predicted by STRING network analysis of genes with a 3′UI in proximal position in zebrafish, mouse and human. Nodes are colored by gene/protein function: nervous system (red), presence of RNA recognition motif (RRM) (green), diseases of signal transduction (blue), FoxO signaling pathway (yellow). (167 nodes and 127 edges in total, PPI enrichment p-value = 0.02). B. Gene ontology enrichment analysis of all 167 genes with conserved 3′UI proximal positioning. The most significant GO term within the following functional categories are shown: Interpro domains, Biological process and Reactome pathways. C. A scatter plot showing fold changes for APPRIS-annotated proximal 3′UI transcripts (dark blue: FC > 1.5 and light blue: FC < 1.5) and distal 3′UI transcripts (black: FC > 1.5 and gray: FC < 1.5) in |
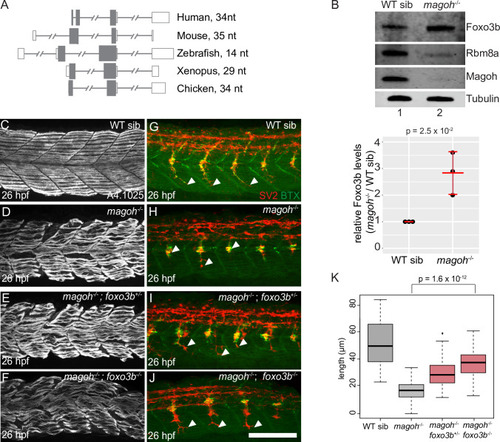
A. Illustration showing EXPRESSION / LABELING:
PHENOTYPE:
|
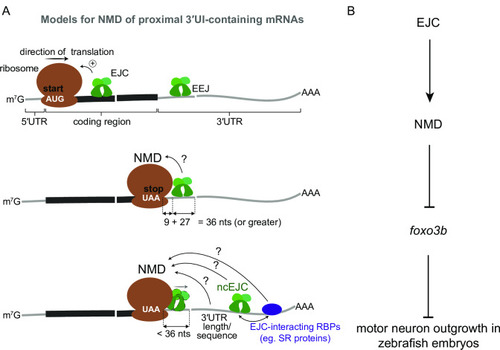
A. Top: EJCs can function to enhance NMD via translation stimulation. Ribosome (brown), direction of translation (straight arrow), start codon (‘AUG’ in white), EJC (green), exon-exon junction (EEJ), mRNA coding region (thick black line) and mRNA 5′ and 3′UTRs (thin gray lines) are labeled. Middle: a model for downstream EJC function in NMD of transcripts where the distance between stop codon (‘UAA’ in white) and downstream exon-exon junction is 36 nts or greater to accommodate both the EJC and the ribosome. Bottom: a model for NMD of transcripts where the distance between stop codon and downstream exon-exon junction is less than 36 nts. Displacement of stop codon-proximal EJC by the ribosome is shown. In this case other downstream factors such as a non-canonical EJC (ncEJC), EJC-interacting RBPs (e.g. SR proteins), or 3′UTR length and/or specific sequences may be responsible for NMD. B. A schematic depicting a model for EJC- and NMD-dependent regulation of |
|
A. Multiple sequence alignments of Eif4a3, Rbm8a and Magoh protein sequences from organisms on the left. Consensus sequence is at the bottom with upper case letters indicating identity and lower case letters indicating similarity. Green indicates complete identity across all species, yellow and blue indicate the identical and unique amino acids in the regions with similarity. Identity between human and zebrafish EJC proteins: Eif4a3 (97%), Rbm8a (93%) and Magoh (100%). B. Western blot detecting proteins listed on the left in RNase I-treated zebrafish embryo total extract (TE, lane 1), depleted extract (DE, lanes 2, 4 and 6) and immunoprecipitates (IP, lanes 3, 5 and 7) with the Rbm8a antibody. Detergents supplemented to increase IP stringency are indicated on top of each lane. Optimized IP condition used in S1C is indicated by the dashed red box. C. Autoradiogram of γ32P 5′-end labeled RNAs from anti-Rbm8a RIP elution (lane 4) as well as indicated size-markers which include the low-molecular weight single-stranded DNA ladder (lane 1), 0.1 pmol 28 nt synthetic RNA (lane 2) and 100 bp DNA ladder (lane 3). D. Scatter plots comparing read counts for each gene in a pair of RIP-Seq replicates. The replicates (Rep1, Rep2, and Rep3) are indicated on the x- and y-axes. A pseudocount of 0.0001 was added to all genic read counts before log2 transformation. Pearson correlation coefficient (r) and p-value for the correlation test for each comparison is on the top left of each plot. E. Genome browser screenshots showing read coverage of Rbm8a RIP-Seq (only Rep 3, the deepest replicate is shown) in green and RNA-Seq in gray of select highly-expressed genes, krt4, eef2b, eif4g1a, and hist1h4l (intron-less gene). |
|
A. Whole mount images of live 19 hpf EJC mutant embryos and WT sibling embryos stained with acridine orange. B. Whole mount images of live EJC mutant embryos and WT siblings at 21 hpf. C. Whole mount images of live EJC mutant embryos and WT siblings at 27 hpf. D. Whole mount images of live EJC mutant embryos and WT siblings at 32 hpf. Head necrosis is indicated by the dashed circle. |
|
A. Venn diagram showing the overlap of genes that are significantly upregulated in EJC mutant embryos and upf1 morphant embryos at 24 hpf [55]. Hypergeometric test p-values for each comparison are also shown. B. MA plot showing the genes that are altered in expression (fold change > 1.5 and FDR < 0.05 in red and unchanged genes in gray) in upf1 morphant embryos compared to control embryos at 12 hpf. The number of significantly upregulated genes is at the top right and the number of downregulated genes is at the bottom right. C. Venn diagram showing the overlap of significantly upregulated genes in upf1 morphant embryos at 24 hpf [55] and upf1 morphant embryos at 12 hpf. Hypergeometric test p-value for the comparison is indicated. D. Empirical CDF plot showing the fold changes in upf1 morphant embryos (24 hpf) [55] of upregulated (blue) and unchanged genes (black) in 21 hpf magoh mutant embryos. Kolmogorov-Smirnov (KS) test p-value for differences in the two distributions are indicated at the bottom of the class descriptions. E. Empirical CDF plot as in S3D for upregulated (red) and unchanged genes (black) in rbm8a mutant embryos at 21 hpf. F. Empirical CDF plot as in S3D for upregulated (red) and unchanged genes (black) in rbm8a mutant embryos at 27 hpf. G. Empirical CDF plot showing the fold changes in upf1 morphants (12 hpf) of upregulated (red) and unchanged genes (black) in rbm8a mutant embryos at 21 hpf. H and I. Proportion of uORF-containing (H) or 3′UI-containing (I) genes in the total number of genes showing significant (FDR < 0.05) fold changes in rbm8a mutant, magoh mutant and upf1 morphant embryos. Genes are divided into four categories based on their log2 fold change: >1.5, 1.5 to 0, 0 to -1.5 and < 1.5. |
|
A. Histogram depicting the frequency of all zebrafish 3′UI transcripts in Ensembl GRCz10 (with APPRIS annotation) as a measure of the distance of the 3′UI from the stop codon. Data are shown in 5 nts bins and bins beyond 500 nts are not shown. Bins of proximal 3′UI genes are in blue and distal 3′UI bins are in gray. Inset: Histogram of all zebrafish proximal 3′UI transcripts binned by 1 nt. B. PANTHER14.0 [87] gene ontology (GO) term enrichment analysis of proximal 3′UI-containing genes (top, shades of blue) and all 3′UI-containing genes (bottom, shades of gray). All significant terms (Benjamini-Hochberg corrected p-value < 0.05) are shown for each set. C. A scatter plot showing gene-level fold change (FC) for transcripts with proximal 3′UI (dark blue: FC > 1.5 and light blue: FC < 1.5) and distal 3′UI (black: FC > 1.5 and gray: FC < 1.5) in rbm8a mutant embryos at 21 hpf compared to wild-type siblings. Genes encircled in red also contain a uORF as determined from a previously published dataset (see Materials and Methods). D. A scatter plot as in C showing fold changes of 3′UI-containing genes for 12 hpf upf1 morphants compared to wild-type control embryos. E. Integrated genome browser (IGV) screenshots of Sashimi plots showing RNA-seq reads observed for foxo3b, phlda2 and cdkn1ba in four zebrafish 24 hpf whole embryo RNA-seq datasets (as labeled on figure in different colors) obtained from the DanioCode consortium. Range of the number of reads mapping to the genes are indicated to the left of each track in black. Number of junction reads are indicated at the spliced junction in the color corresponding to the specific track. In case of foxo3b, due to the length of the second intron the screenshots of the first two and the last two exons are shown separately. F. Empirical CDF plot showing the fold changes in magoh mutants (21 hpf) of genes upregulated that contain a proximal (green) or distal (light purple) 3′UI or intron-less genes (black). Genes that also contained an uORF were excluded from analysis. Kolmogorov-Smirnov (KS) test p-value for differences in fold changes between the two groups is indicated on the bottom right. G. CDF plot as in F showing the fold changes in rbm8a mutants (27 hpf) of genes upregulated that contain a proximal (green) or distal (light purple) 3′UI or intron-less genes (black). Kolmogorov-Smirnov (KS) test p-value for differences in fold changes between the two groups is indicated on the bottom right. |
|
A. Histogram showing the frequency of all APPRIS 3′UI-containing transcripts in human GRCh38 as a measure of the distance of the 3′UI from the stop codon. Data are grouped in 5 nt bins from 1–500 nts. Proximal 3′UI-containing gene bins are indicated in blue; distal 3′UI-containing gene bins are indicated in gray. Red dotted line indicates distance from stop codon to farthest 3′UI = 50 nts. Histogram of transcripts from Ensembl annotation is shown in C. B. Histogram as in A of mouse proximal and distal 3′UI-containing transcripts in mouse GRCm38. Histogram of transcripts from Ensembl annotation is shown in D. C. Histogram as in A of proximal and distal 3′UI-containing human transcripts from Ensembl annotation of GRCh38. D. Histogram as in A of proximal and distal 3′UI-containing mouse transcripts from Ensembl annotation of GRCm38. E. PANTHER14.0 [86] gene ontology (GO) term enrichment analysis of human APPRIS proximal 3′UI-containing genes (shades of blue) and all human APPRIS 3′UI-containing genes (shades of gray). All significant terms (Benjamini-Hochberg corrected p-value < 0.05) are shown for each set. F. GO term enrichment analysis as in E of mouse APPRIS 3′UI-containing genes. G. A scatter plot showing gene-level fold changes for all Ensembl-annotated transcripts with proximal 3′UI (dark blue: FC > 1.5 and light blue: FC < 1.5) and distal 3′UI (black: FC > 1.5 and gray: FC < 1.5) in UPF1 knockdown human embryonic kidney cells (HEK293) compared to control cells using previously published data [57]. Genes encircled in red also contain a uORF as determined from a previously published dataset (see Methods). H. A scatter plot showing gene-level fold changes for all APPRIS-annotated transcripts with proximal 3′UI (dark blue: FC > 1.5 and light blue: FC < 1.5) and distal 3′UI (black: FC > 1.5 and gray: FC < 1.5) in UPF1 knockdown human embryonic stem cells (hESCs) compared to control cells using previously published data [62]. Genes encircled on red also contain a uORF as determined from a previously published dataset (see Materials and Methods). I. Scatter plot as in E showing gene-level fold changes of APPRIS-annotated mouse 3′UI-containing transcripts in Smg6-/- knockout mouse embryonic stem cells (mESCs) compared to wild-type cells [63]. J. Western blots showing representative protein knockdown in one replicate for Fig 7E. |
|
A. Semi-quantitative RT-PCR shows transcript levels of foxo3b, eif4a2 and rpl13 (loading control) in rbm8a mutant and wild-type sibling embryos at 21 and 27 hpf. B. Western blots (on the left) show levels of Foxo3b, Rbm8a, Magoh, and Tubulin in rbm8a mutant embryos (lane 2) compared to WT siblings (lane 1) at 27 hpf (N = 20 embryos per genotype). Right: dot plot showing Foxo3b levels normalized to tubulin levels in rbm8a mutant embryos and WT siblings at 27 hpf in three biological replicates. Error bars: standard error of means. Welch’s t-test p-values are indicated at the top. C. Bar graph showing log2 fold changes of known Foxo3b transcriptional targets that show a significant upregulation (FDR < 0.05) in EJC mutant RNA-Seq datasets. Foxo3b targets are from Morris et al. 2015 [64]. A pound symbol indicates statistically non-significant log2 fold change with FDR > 0.05. D-G. Confocal images showing Myh1 immunofluorescence using anti-A4.1025 in somites 12–16 of WT sibling (D), rbm8a-/- mutant (E), rbm8a-/-; foxo3b+/- mutant (F), and rbm8a-/-; foxo3b-/- mutant (G) embryos. (N = 5 embryos/genotype). H-K. Merged confocal images showing motor neurons (red; detected by anti-SV2 staining) and acetylcholine receptors (green; detected by alpha-bungarotoxin staining) in somites 12–16 of WT sibling (H), rbm8a-/- mutant (I), rbm8a-/-; foxo3b+/- mutant (J), and rbm8a-/-; foxo3b-/- mutant (K) embryos. Neuromuscular junctions in the merged image are yellow. White arrowheads point to the distal end of the motor neuron. (N = 5 embryos/genotype). Scalebar in K (for panels D-K) is 100 nm. L. Boxplots showing quantification of motor axon length in embryos of genotypes indicated along the x-axis) (4 motor neurons/embryo and 5 embryos/genotype). Welch’s t-test p-values are indicated at the top. |

Unillustrated author statements |