
- Title
-
RRM2 enhances MYCN-driven neuroblastoma formation and acts as a synergistic target with CHK1 inhibition
- Authors
- Nunes, C., Depestel, L., Mus, L., Keller, K.M., Delhaye, L., Louwagie, A., Rishfi, M., Whale, A., Kara, N., Andrews, S.R., Dela Cruz, F., You, D., Siddiquee, A., Cologna, C.T., De Craemer, S., Dolman, E., Bartenhagen, C., De Vloed, F., Sanders, E., Eggermont, A., Bekaert, S.L., Van Loocke, W., Bek, J.W., Dewyn, G., Loontiens, S., Van Isterdael, G., Decaesteker, B., Tilleman, L., Van Nieuwerburgh, F., Vermeirssen, V., Van Neste, C., Ghesquiere, B., Goossens, S., Eyckerman, S., De Preter, K., Fischer, M., Houseley, J., Molenaar, J., De Wilde, B., Roberts, S.S., Durinck, K., Speleman, F.
- Source
- Full text @ Sci Adv
|
|
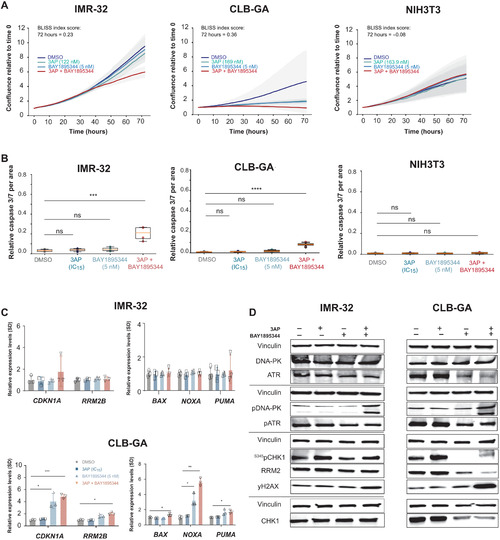
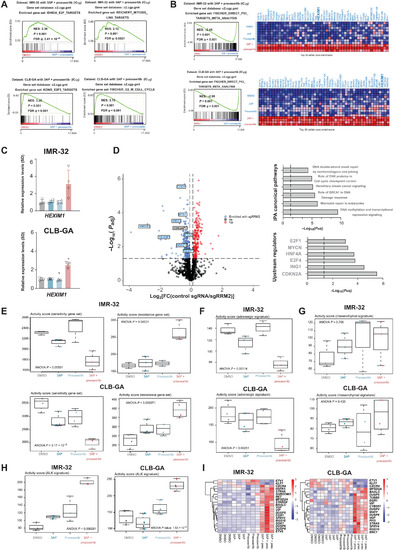
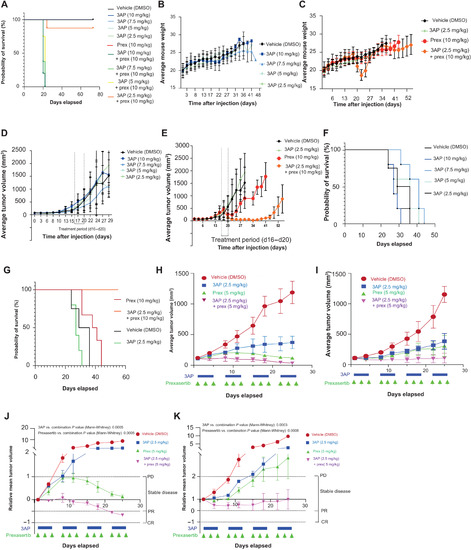
PHENOTYPE:
|
|

|
|

|
|
|