
- Title
-
Analysis of zebrafish periderm enhancers facilitates identification of a regulatory variant near human KRT8/18
- Authors
- Liu, H., Duncan, K., Helverson, A., Kumari, P., Mumm, C., Xiao, Y., Carlson, J.C., Darbellay, F., Visel, A., Leslie, E., Breheny, P., Erives, A.J., Cornell, R.A.
- Source
- Full text @ Elife
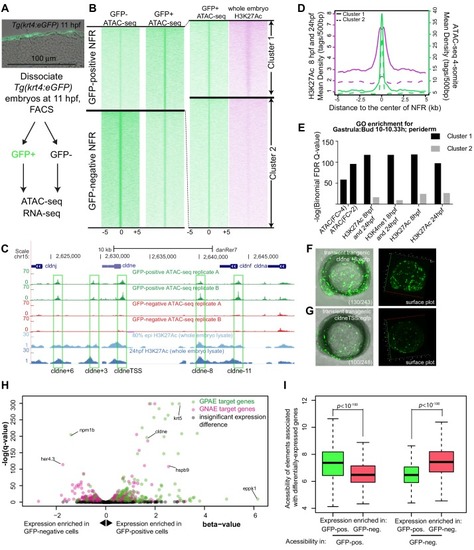
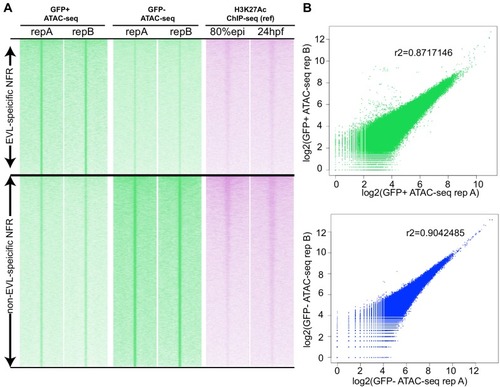
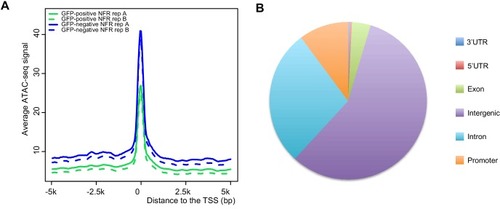
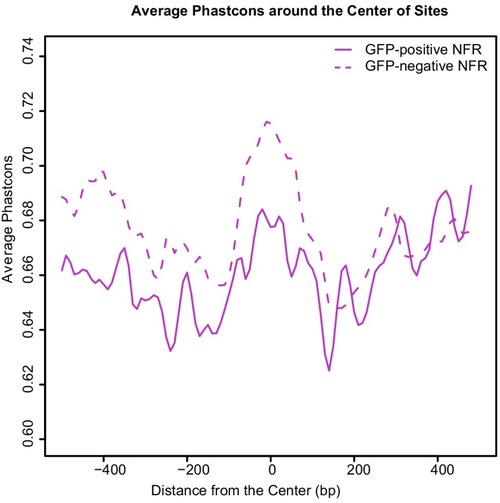
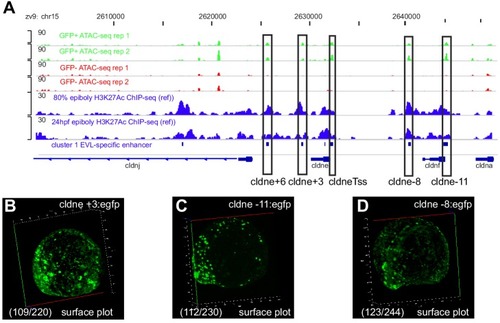
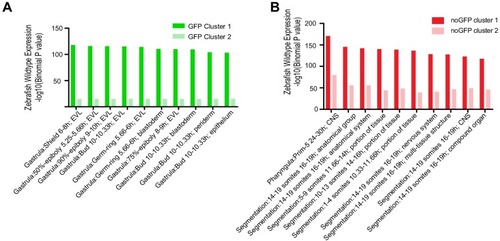
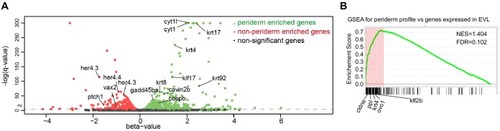
(A) Transverse section of an 11 hpf (4-somite stage) Tg(krt4:gfp) embryo, showing GFP is confined to the superficial layer of cells, and workflow of ATAC-seq in periderm and non-periderm cells. (B) Density plots of ATAC-seq results. Each line is centered on a nucleosome free region (NFR) with significantly more ATAC-seq reads in GFP-positive or GFP-negative cells; the majority of ATAC-seq peaks were not enriched in either cell type. Density plots also show H3K27Ac ChIP-seq signal in whole embryos at eight hpf and/or at 24 hpf data from Bogdanovic et al. (2012) at each of the GFP-positive NFRs; the latter are sorted in to those that overlap (or are flanked by within 100–1500 bp) peaks of H3K27Ac signal (cluster 1, 4301 elements) and or not (cluster 2, 7952 elements). (C) UCSC Genome browser tracks showing the ATAC-seq peaks in GFP-positive and GFP-negative cells, and H3K27Ac signal from whole embryos at eight hpf and at 24 hpf data from Bogdanovic et al. (2012) at the cldne locus. Boxes, examples of cluster one elements, also known as zebrafish GFP-positive active enhancers (zGPAEs). Elements are cldne+6 kb (zv9 : chr15:2625460–2625890), cldne +3 kb (chr15:2629012–2629544), cldne −8 kb (chr15:2639873–2640379), cldne −11 kb (chr15:2643578–2644160), and cldne TSS (chr15:2631981–2632513). (D) Plot of average density of H3K27Ac ChIP-seq signal (purple) and ATAC-seq signal (green). (E) GO enrichment for term ‘Gastrula:Bud 10–10.33 hr; periderm’ among NFRs enriched in GFP-positive cells with normalized fold change greater than 2 (ATAC(FC >2)) and 4 (ATAC(FC >4)), NFRs enriched in GFP-positive cells flanked or overlapped by 24hpf and 80% epiboly H3K27Ac ChIP-seq peaks (cluster 1) and or not (cluster 2), NFRs enriched in GFP-positve cells flanked or overlapped by 24hpf and 80% epiboly H3K4me1 ChIP-seq peaks (cluster 1) or not(cluster 2), NFRs enriched in GFP-positive cells flanked or overlapped by 24hpf H3K27Ac ChIP-seq peaks (cluster 1) or not (cluster 2), and NFRs enriched in GFP-positive cells flanked or overlapped by 80% epiboly H3K27Ac ChIP-seq peaks (cluster 1) or not (cluster 2). (F), (G) Lateral views of wild-type embryos at 11 hpf injected at the 1-cell stage with GFP reporter constructs built from (F) cldne +6 and (G) cldne transcription start site (TSS) elements. Left panels are stack views of the embryo, and right panels are surface plot for the embryos indicating most GFP signal is from the surface (periderm) of the embryos. Number in parentheses is the ratio of embryos with at least 10 GFP-positive periderm cells over injected embryos surviving at 11 hpf. (H) Volcano plot of RNA seq data, showing the expression of genes associated (by GREAT) with zGPAEs (green dots) or with zGNAEs (pink dots) in GFP-positive cells (beta-value >0) or in GFP-negative cells (beta-value <0). (I) Plot of accessibility scores of elements with differential accessibility (i.e., both zGPAEs and zGNAEs) associated with genes that are differentially expressed in GFP-positive and GFP-negative cells, showing that elements with increased accessibility in GFP-positive cells tend to be associated with genes whose expression is enriched in GFP-positive cells, and vice versa. EXPRESSION / LABELING:
|
( |
( |
|
( |
( |
( |

|

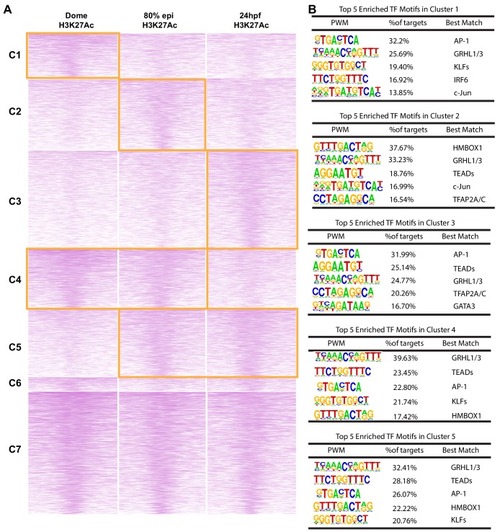
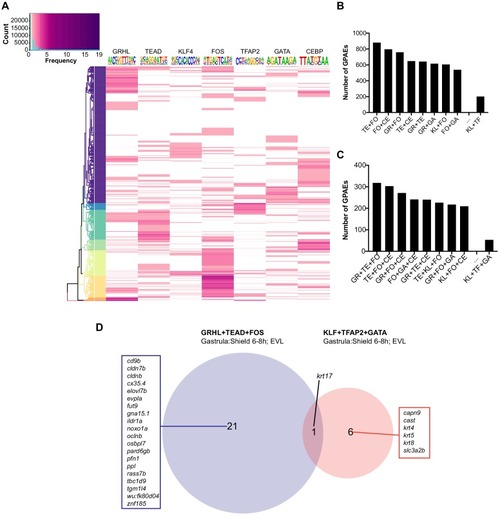
(A) Enriched motifs in zGPAEs. PWM, position weighted matrix. TF, transcription factors. Best match, transcription factor in the indicated family with highest expression in GFP-positive cells, whether or not the expression is enriched in GFP-positive cells in comparison to GFP-negative cells. (B) Genome browser view showing a GFP-positive nucleosome free region (NFR) about 3 kb downstream of the transcription start site of cldne gene. (C) Schematic of frequency of Tn5 cleavage sites at within this NFR, indicating reduced frequency of cleavage at a motif matching the GRHL binding site relative to in flanking DNA. (D) Confocal image of a wild-type embryo at 10 hpf (2-somite stage) injected at the one-cell stage with a reporter construct containing this NFR. (E) Bar chart showing number of embryos positive for GFP signal in the periderm after being injected with the intact reporter or one in which the GRHL motif was deleted. (F) Bar chart showing the percentage of genes whose expression is higher in GFP-positive cells than in GFP-negative cells that are flanked by a zGPAE possessing the indicated binding site. |
( |

|

Depending on the expression level in periderm cells (GFP-positive cells) most enriched transcription factors with the relevant motifs are in hexagon while other enriched transcription factors with the relevant motifs are in round. Each TF node is colored according to the normalized expression z-score (related to periderm genes). The thickness of each edge represents the number of motifs located in the all nearby enhancers to each TF (within 100kbp to the transcription start site). |
( |

GO enrichment assay of gene expression for the top-scoring 10 K tiles that do not overlap zGPAEs. |
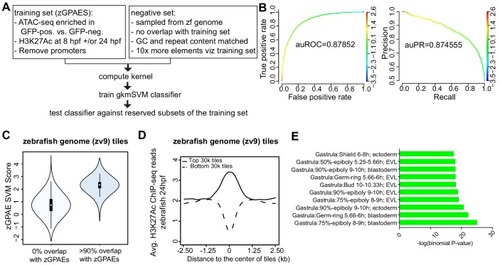
(A) Pipeline for training and cross-validation of gkmSVM classifier on zebrafish periderm enhancer candidates. (B) Receiver Operating Characteristic (ROC) and Precision-Recall (PR) curves using the gkmSVM trained on zGPAEs. au, area under. Color of curves corresponds to SVM scores. (C) Violin plots showing SVM scores of zebrafish genome tiles with 0% or at least 90% overlapped with the training set (GPAEs). (D) Average H3K27Ac ChIP-seq reads at the 30,000 elements with the highest or lowest scores from the gkmSVM trained on zGPAEs. (E) GO enrichment assay for genes associated with the top-scoring tiles 10,000 tilesincluding those that overlap the training set. |
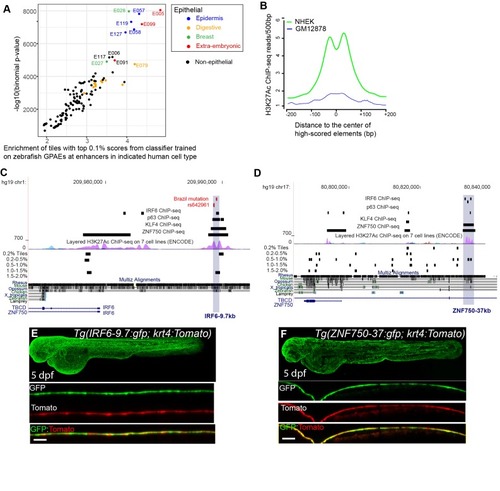
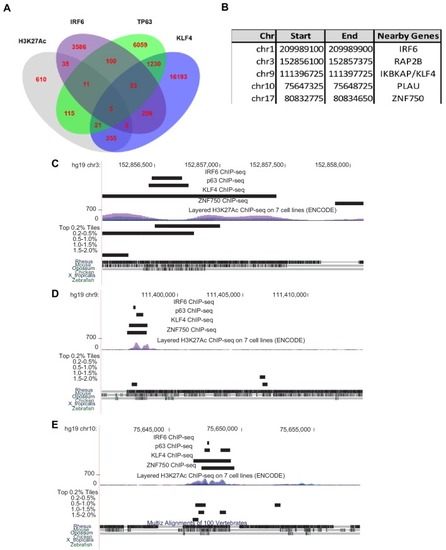
(A) Enrichment of human genome tiles that receive a top 0.1% bin score using a zGPAE-trained classifier at enhancers active in the indicated cell type, as revealed by ChIP-seq to chromatin marks in the Roadmap Epigenomics project (Visel et al., 2008). -Such tiles are significantly enriched within a variety of epithelial enhancers. [E05: H1 BMP4 Derived Trophoblast Cultured Cells; E027: Breast Myoepithelial Primary Cells; E028: Breast variant Human Mammary Epithelial Cells; E057, E058: Foreskin Keratinocyte Primary Cells; E079: Esophagus; E091: Placenta; E099: Placenta amnion; E119, Mammary Epithelial Primary Cells (HMEC); E127:NHEK-Epidermal Keratinocyte Primary Cells]. (B) Average density of H3K27Ac ChIP-seq signal in NHEK and GM12878 cells (Visel et al., 2008) at top 0.1% tiles using a zGPAE-trained classifier. (C) Genome browser view focused on IRF6-9.7, also known as IRF6 multispecies conserved sequence 9.7 (MCS9.7) (hg19 chr1:209989050–209989824). A SNP within it, rs642961 (chr1: 209989270), is associated with risk for non-syndromic orofacial cleft. Brazil mutation refers to a rare mutation reported in a patient with Van der Woude syndrome (Fakhouri et al., 2012). This element has peaks of H3K27Ac, IRF6, TP63, and KLF4 ChIP-seq in normal human keratinocytes. Multiz Alignments of 100 vertebrate species shows it is conserved among mammals but not in zebrafishNonetheless it possesses tiles in the top 1.0-1.5% and 0.2% bins using a zGPAE-trained classifier (D) Genome browser view focused on ZNF750-37 (hg19 chr17:80832267–80835105). This element has similar ChIP-seq signature as IRF6-9.7, and like it is not overtly conserved to fish but posseses high-scoring tiles using the zGPAE-trained classifier. (E) GFP expression pattern of Tg(IRF6-9.7:gfp; krt4:Tomato) at five dpf. (F) GFP expression pattern of Tg(ZNF750-37:gfp; krt4:Tomato) at five dpf. Both of these human non-coding elements have periderm enhancer activity in zebrafish embryos. EXPRESSION / LABELING:
|

( EXPRESSION / LABELING:
|
( |

Transient transgenic reporter assays for enhancer candidates near human PPL and zebrafish ppl. ( |
( |
( |
( |
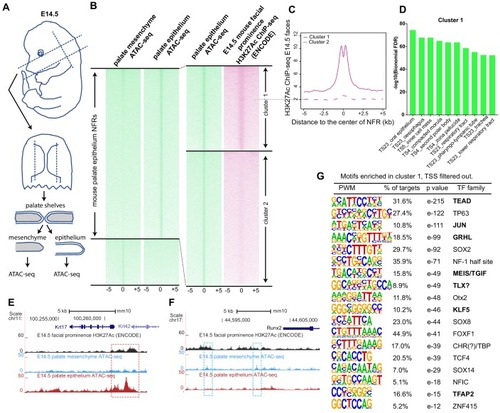
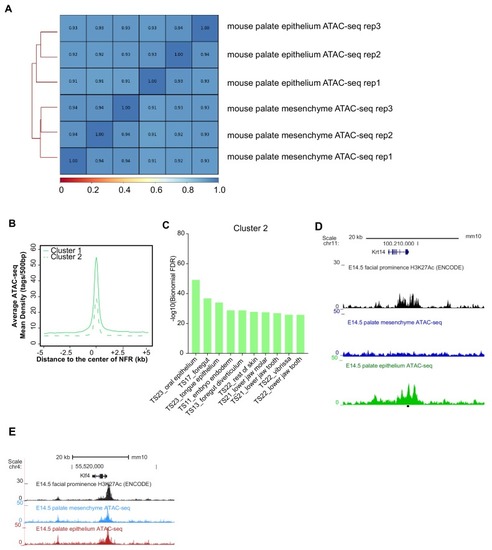
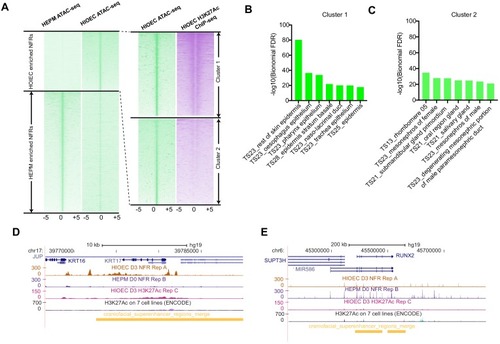
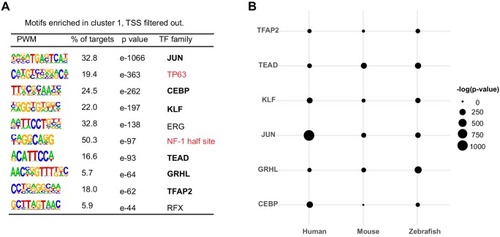
(A) Workflow of ATAC-seq in epithelium and mesenchyme cells isolated from palate shelves dissected from E14.5 embryos. (B) Heatmap plots of ATAC-seq and E14.5 mouse facial prominence H3K27Ac ChIP-seq (Klein and Andersen, 2015) in tissue-specific NFRs. (C) Plot of average density of H3K27Ac ChIP-seq signal, showing higher signal at cluster 1 elements than cluster 2 elements. (D) GO enrichment (MGI mouse gene expression pattern) of genes associated with cluster 1 elements. (E and F) UCSC Genome browser views of the mouse genome (mm10 build) showing the ATAC-seq and H3K27Ac ChIP-seq signals near the Krt17 and Runx2 genes. Red box, an example of a mouse palate-epithelium active enhancer (mPEAE). Blue boxes, examples of mouse palate mesenchyme active enhancers (mPMAEs). (G) Motifs enriched in cluster 1 of E14.5 palate-epithelium specific NFRs with elements overlying transcription start sites removed (i.e., mPEAEs). Motifs shared with zGPAEs are in bold. |
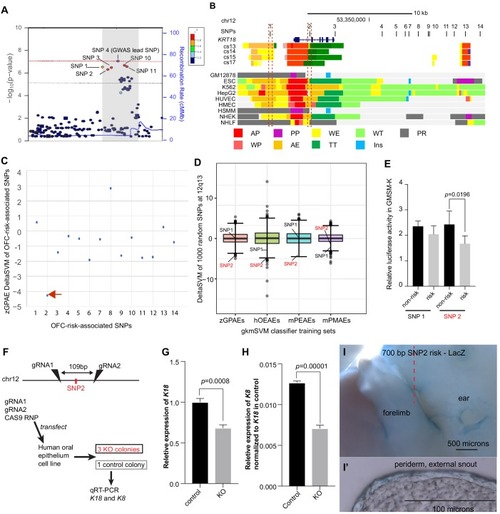
(A) Regional plot showing OFC-risk-associated single nucleotide polymorphism (SNPs) near KRT18 from this study. SNP4 is the lead SNP from our meta-analysis of OFC GWAS (Leslie et al., 2017). (B) Browser view of the human genome, hg19, focused on this locus. Tracks: SNPs: OFC-risk-associated SNPs. SNP1: rs11170342, SNP2: rs2070875, SNP3: rs3741442, SNP4: rs11170344, SNP5: rs7299694, SNP6: rs6580920, SNP7: rs4503623, SNP8: rs2363635, SNP9: rs2682339, SNP10: rs111680692, SNP11: rs2363632, SNP12: rs4919749, SNP13: rs2638522, SNP14: rs9634243. Color coded bars: Chromatin status (color code explained in key), revealed by ChIP-seq to various chromatin marks. Cs13-cs17, facial explants from human embryos at Carnegie stage (cs) 13–17, encompassing the time when palate shelves fuse (Wilderman et al., 2018). Roadmap Epigenomics Project cell lines (Visel et al., 2008): GM12878, B-cell derived cell line; ESC, Embryonic stem cells; K562, myelogenous leukemia; HepG2, liver cancer; HUVEC, Human umbilical vein endothelial cells; HMEC, human mammary epithelial cells; HSMM, human skeletal muscle myoblasts; NHEK, normal human epidermal keratinocytes; NHLF, normal human lung fibroblasts. AP, active promoter; WP, weak promoter; PP, poised promoter; AE, active enhancer; WE, weak enhancer; TT, transcriptional transition; WT, weakly transcribed; Ins, insulator; PR, polycomb-repressed. (C) deltaSVM scores predicted by zGPAEs-derived classifier for the 14 OFC associated SNPs near KRT18. (D) Box and whisker plots of deltaSVM scores of 1000 randomly-selected SNPs near KRT18, scored by classifiers trained by zGPAEs (zebrafish periderm active enhancers), hOEAEs (human oral epithelium active enhancers), mPEAEs (mouse palatal epithelium active enhancers) and mPMAEs (mouse palatal mesenchyme active enhancers). The line is the median scoring SNP, the box contains the middle-scoring two quartiles, and the whisker represent the top and lower quartiles. Dots are outliers. deltaSVM scores for SNP1 and SNP2 are indicated. Number out of 1000 randomly selected SNPs with a lower deltaSVM than SNP2 with classifier trained on zGPAEs, 2; on mPEAEs, 9; on hOEAEs, 17; on mPMAEs, 186. (E) Dual luciferase assay for non-risk and risk alleles of rs11170342 (SNP1) and rs2070875 (SNP2) in GMSM-K cells. (F) Schematic diagram showing the workflow of generating GMSM-K cell colonies with 109 bp flanking SNP2 deleted by CRISPR-Cas9. (G,H) qRT-PCR showing relative RNA expression of KRT18 (G) and KRT8 (H) in three homozygous knockout colonies (KO) and one isolated wild-type colony (Control) of GMSM-K cell lines. (I) Lateral view of transgenic mice LacZ reporter assay for the 700 bp DNA fragment overlapping SNP2. (I’) Section of the facial prominence from I (red circled region). |

Dot plot of deltaSVM scores for each SNP calculated with classifiers trained on the indicated set of enhancer candidates |
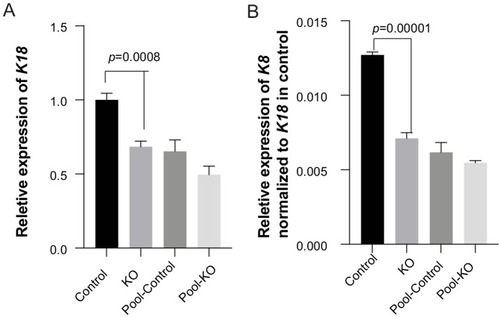
KO: three homozygous knockout colonies; Control: one isolated wildtype colony; Pool-control: pool of GMSM-K cells transfected with two gRNAs only; Pool-KO: Pool of GMSM-K cells transfected with two gRNA along with Cas9 RNP. |

( |