
- Title
-
Conformational dynamics modulate the catalytic activity of the molecular chaperone Hsp90
- Authors
- Mader, S.L., Lopez, A., Lawatscheck, J., Luo, Q., Rutz, D.A., Gamiz-Hernandez, A.P., Sattler, M., Buchner, J., Kaila, V.R.I.
- Source
- Full text @ Nat. Commun.
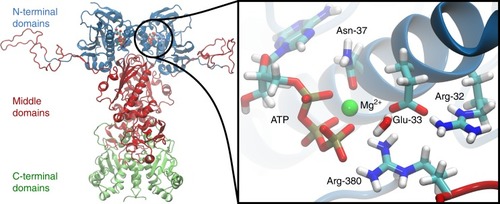
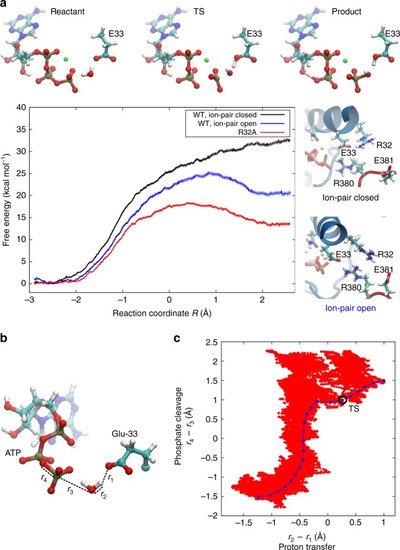
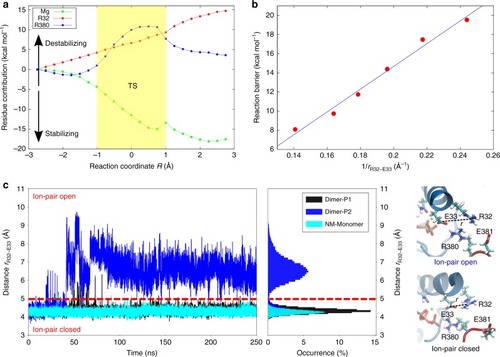
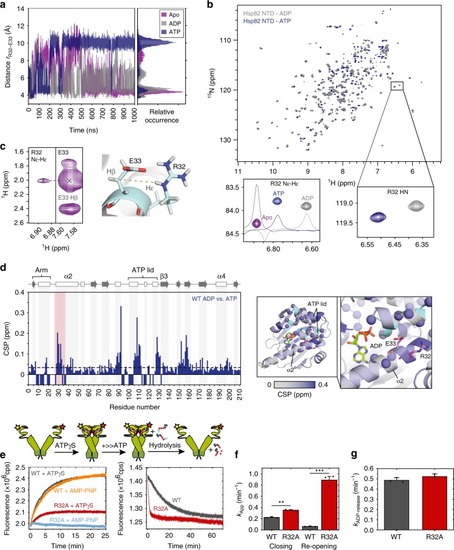
The N-terminal domains of Hsp90 are shown in blue, the middle domains in red, and the C-terminal domains in green. The inset shows a structure of the active site with a bound ATP molecule obtained from an MD simulation, where Asn-37 undergoes a rotation to form a stronger coordination to the magnesium. |
|
|
|
|