
- Title
-
Leveraging chromatin state transitions for the identification of regulatory networks orchestrating heart regeneration
- Authors
- Cordero, J., Elsherbiny, A., Wang, Y., Jürgensen, L., Constanty, F., Günther, S., Boerries, M., Heineke, J., Beisaw, A., Leuschner, F., Hassel, D., Dobreva, G.
- Source
- Full text @ Nucleic Acids Res.

H3K27ac dynamics during zebrafish heart regeneration. ( |
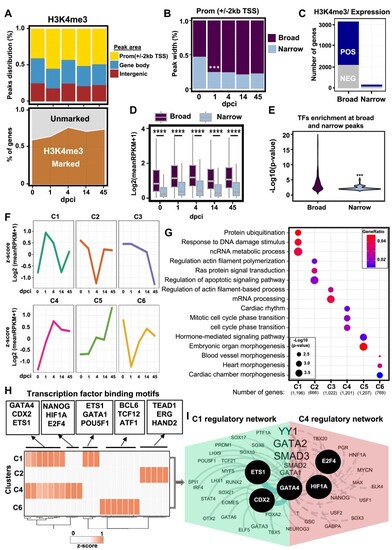
H3K4me3 breadth correlates with transcriptional activity in zebrafish heart regeneration. ( |

Large-scale acquisition of repressive chromatin marks one day after heart injury. ( |

Switch between inactive to active chromatin states four days after injury. ( |

Transition from active to inactive chromatin state between 4 dpci and 14 dpci marks the start of the healing process. ( |

Conservation of chromatin state transitions between zebrafish and neonatal mice after heart injury. ( |

Functions of identified TFs in CM dedifferentiation and proliferation. ( |