
- Title
-
Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart
- Authors
- Bendig, G., Grimmler, M., Huttner, I.G., Wessels, G., Dahme, T., Just, S., Trano, N., Katus, H.A., Fishman, M.C., and Rottbauer, W.
- Source
- Full text @ Genes & Dev.

Effects of the msq m347 mutation on heart function, morphology, and expression of anf. (A,B) Lateral view of wild-type (wt) (A) and msq mutant embryos (B) at 72 hpf. msq mutants develop pericardial edema due to loss of ventricular contractility, whereas the development of other organ systems proceeds normally. (C–F) RNA levels of the stretch-responsive gene zanf are severely reduced in msq mutant hearts. In contrast to wild-type hearts, where robust expression of zanf throughout the heart can be observed at 48 hpf (C) and 72 hpf (D), zanf expression in msq mutant hearts is only slightly reduced at 48 hpf (E), but completely absent by 72 hpf (F). Atrium (A) and ventricle (V) are indicated. (G,H) Contractility of msq mutant zebrafish embryo ventricles severely decreases over time; by 96 hpf, the ventricular chamber becomes completely silent. Measurements of fractional shortening (FS)—an indicator of systolic contractile function normalized to the diameters of the heart—are displayed for the ventricular chamber of wild-type zebrafish embryos (G) and msq mutants (H) at different time points. (I,J) The msq mutation does not affect overall heart morphology. Hematoxylin/eosin-stained histological sections of wild-type (I) and msq mutant (J) heart at 72 hpf. Endocardial (en) and myocardial (my) layers of ventricle and atrium of msq mutant heart are unaltered. (K,L) Transmission electron microscopy of wild-type (K) and msq mutant (L) zebrafish embryonic hearts at 72 hpf. Ventricular cardiomyocytes of msq mutants show normal cell architecture and organization of myofibrils. Insets display transverse sections of myofilaments. EXPRESSION / LABELING:
PHENOTYPE:
|
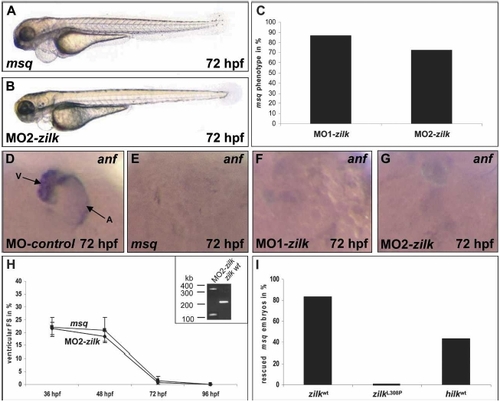
Antisense oligonucleotide-mediated knockdown of zilk phenocopies the msq mutant heart phenotype, whereas ectopic expression of wild-type ILK suppresses cardiac dysfunction in msq mutants. (A–H) Inhibition of zilk function by morpholino-modified antisense oligonucleotide injection phenocopies the msq mutant phenotype. MO2-zilk injected embryos (B) are indistinguishable from msq mutant embryos (A) and display severe impairment of ventricular contractility (H). (C) Eighty-seven percent of MO1-zilk-injected, and 72.1% of MO2-zilk-injected wild-type embryos display the msq mutant phenotype at 72 hpf. (D–G) Similar to the situation in msq mutant hearts (E), injection of either MO1-zilk (F) or MO2-zilk (G) leads to reduced zanf RNA levels in the heart. (D) In contrast, injection of MO-control has no effect on zanf expression. Atrium (A) and ventricle (V) are indicated. (H) Ventricular fractional shortening (FS) of MO2-zilk-injected and msq mutant embryos. The effect of MO2-zilk on mRNA splicing is shown in the inset. Injection of MO2-zilk results in abnormal splice products of 380 bp (integration of intron 4) and 125 bp (skipping of exon 4), predicted to lead to premature termination of translation of zilk. (I) Ectopic expression of wild-type ilk RNA from either zebrafish (zilk wt) or human (hilk wt) can rescue the heart phenotype of a significant proportion of msq mutant embryos, whereas injection of msq mutant RNA (zilk L308P) has no effect. EXPRESSION / LABELING:
PHENOTYPE:
|
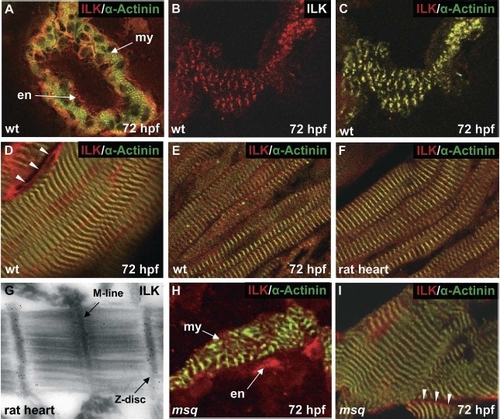
ILK localizes to the sarcolemma and sarcomeric Z-discs of cardiomyocytes. (A) ILK is expressed in the endocardial (en) and myocardial (my) layer of the zebrafish heart. Cross-section through the cardiac ventricle of a wild-type zebrafish embryo at 72 hpf. (B,C) Coimmunostaining with an antibody directed against the sarcomeric Z-disc protein α-Actinin reveals colocalization of ILK and α-Actinin in zebrafish cardiomyocytes. (C) Merge of the anti-ILK and anti-α-Actinin stain. (D) As already observed in cardiomyocytes (C), ILK and α-Actinin also colocalize at sarcomeric Z-discs of zebrafish skeletal muscle cells. In addition, as indicated by arrowheads, sarcolemmal localization of ILK can be observed. (E–G) In the adult zebrafish (E) and rat (F,G) heart, ILK and α-Actinin also colocalize at sarcomeric Z-discs. (G) Immuno-gold staining and transmission electron microscopy of rat heart sections confirms localization of ILK at sarcomeric Z-discs. (H,I) msq mutant ILKL308P is expressed at normal levels in msq mutant zebrafish hearts (H) and skeletal muscle (I), and shows sarcolemmal staining (indicated by arrowheads) and co-localization with α-Actinin. EXPRESSION / LABELING:
|
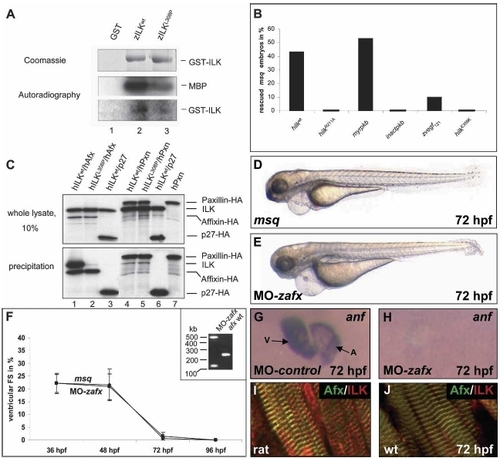
msq mutant ILKL308P disrupts ILK kinase activity and its binding to β-parvin (Affixin). (A) In comparison to zILKwt, the msq mutation zILKL308P leads to potent reduction of phosphorylation of the substrate MBP (middle part), as well as of ILK autophosphorylation (lower part). (Upper part) Identical amounts of purified recombinant zILKwt (lane 2) and zILKL308P (lane 3) were used. Lane 1 shows incubation of MBP with GST as a control. (B) Rescue of the msq mutant heart phenotype by mRNA injection of various constructs. Kinase-deficient hilk (hilk R211A), as well as hilk deficient in β-parvin binding (hilk E359K) fail to rescue the msq mutant phenotype, whereas wild-type hilk wt is able to rescue the msq phenotype. Injection of either zvegf121 mRNA, or myristoylated human PKB (myrpkb) mRNA suppresses the msq mutant phenotype. Injection of kinase inactive hPKB (inactpkb) mRNA has no effect. (C, lower panel) In vitro coimmunoprecipitation with human β-parvin, Paxillin, and ILK. HA-tagged hβ-parvin (hAfx) interacts with wild-type hILKwt (lane 1), but not with mutant hILKL308P (lane 2). In contrast, HA-tagged hPaxillin (hPxn) precipitates with both hILKwt (lane 4) and hILKL308P (lane 5). (Lanes 3,6) As a control, HA-tagged hp27Kip1 is used. Lane 7 indicates precipi-tation of HA-tagged hPaxillin to exclude degradation products of hPaxillin migrating at the size of hILK. The upper panel shows 10% input of the radiolabeled, cotranslated proteins. (D–H) Antisense oligonucleotide-mediated knockdown of zebrafish β-parvin (affixin) phenocopies the msq mutant heart phenotype. MO-zafx injected embryos (E) are indistinguishable from msq mutant embryos (D) and display severe impairment of ventricular contractility. (F) Similar to msq mutant embryos, fractional shortening (FS) of the ventricular chamber decreases significantly in MO-zafx-injected embryos. The effect of MO-zafx on mRNA splicing is shown in the inset. Injection of MO-zafx results in abnormal splice products of 500 bp (integration of intron 5) and 125 bp (skipping of exon 5), both predicted to lead to premature termination of translation of zilk. (G,H) In contrast to wild-type hearts (G), zanf RNA cannot be detected by RNA antisense in situ hybridization in hearts from MO-zafx injected embryos at 72 hpf (H). Atrium (A) and ventricle (V) are indicated. (I,J) β-Parvin (Afx) and ILK colocalize at sarcomeric Z-discs of zebrafish (J) and rat hearts (I). EXPRESSION / LABELING:
PHENOTYPE:
|